[研究成果] 前原班の研究がMol Syst Biol誌に掲載されました!
2021.11.06
Maehara K, Tomimatsu K, Harada A, Tanaka K, Sato S, Fukuoka M, et al. Modeling population size independent tissue epigenomes by ChIL‐seq with single thin sections. Mol Syst Biol. Nov;17(11):e10323 (2021)
組織の細胞集団に潜む幹細胞のエピゲノム解析手法を開発
―がん組織の精密プロファイリングに成功―
【概要】
筋肉や肝臓といった組織は、多様な種類の細胞から構成され、異なる役割を持つ細胞同士が協調的に働くことで、その機能を実現します。組織を構成する細胞の種類は、遺伝子の組み合わせで決定され、エピゲノムがその組み合わせ方を司ると考えられています。これまでの組織のエピゲノム解析は、幹細胞 [用語1] など組織中の少数派細胞の情報が、多数派に埋もれてしまうことが課題となっていました。
九州大学生体防御医学研究所の大川恭行教授、前原一満助教らは、一辺3 mm厚さ10 μm程度の小さな組織切片に含まれる少数の細胞集団から、高感度かつ高精度なエピゲノム [用語2] 情報を抽出する技術を発表しました。この技術は、これまで同グループが発表してきた「クロマチン挿入標識(Chromatin Integration Labeling: ChIL)法(ChIL-seq)」を基礎として、今回さらに組織をターゲットとした解析法を開発しました。
本研究グループは、高感度なChIL-seqの技術と、遺伝子発現を司る分子の移動度を評価する統計モデルを組み合わせ、埋もれた幹細胞の活性化情報を検出する新たな解析技術を開発しました。さらに本技術は、様々なステージの乳がん組織のプロファイリングに有用であることも示しました。この解析技術を使うことで、組織再生や再生医療の応用に必要な幹細胞や、組成が多様ながん組織などの詳細なメカニズムの解明が期待されます。
本研究は、東京工業大学科学技術創成研究院細胞制御工学研究センター(木村宏教授ら)、東京大学定量生命科学研究所(胡桃坂仁志教授ら)、公益財団法人がん研究会がん研究所(斉藤典子部長ら)の研究グループとの共同研究で行われました。
本研究成果は、2021年 11月 3日 正午(中央ヨーロッパ時間) に欧州科学雑誌「Molecular Systems Biology」で公開されました。
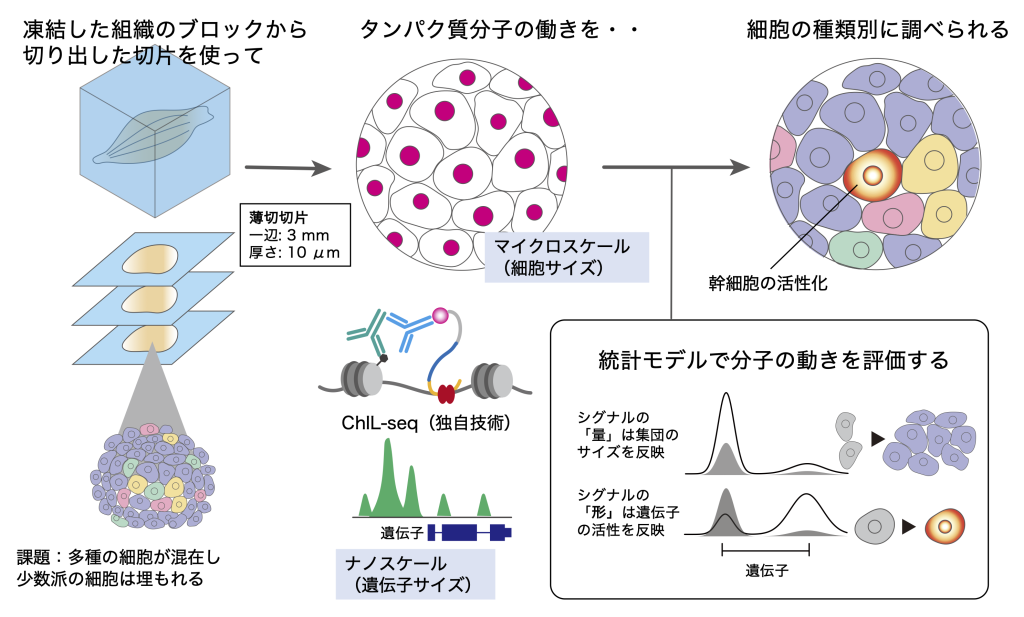 (参考図)研究成果の概要:高感度なChIL-seqと統計モデルを組み合わせ、多数細胞に埋もれない組織のエピゲノム解析を可能にした。
(参考図)研究成果の概要:高感度なChIL-seqと統計モデルを組み合わせ、多数細胞に埋もれない組織のエピゲノム解析を可能にした。
【研究者からひとこと】
計測技術と情報解析技術の一体的な開発が実を結びました。組織エピゲノム解析の課題に対して、我々のChIL-seqのメリットを十全に活かした解決策が提示できたと思います。多くの方にこの技術を使ってもらえると嬉しいです。
【研究成果のポイント】
- 分子の移動度を評価する統計モデルを組み合わせることで、幹細胞のように多数派細胞に埋もれた少数派細胞も含めて組織全体のエピゲノム情報を解析できる新たな解析技術を開発した。
- 3 mm×3 mm×10 μmの小さな組織切片1枚から高感度かつ高精度なエピゲノム情報が取り出せる。
- 従来法より細胞の損失が少なく、細胞にストレスを与えにくい。
【研究の背景】
筋肉や肝臓といった組織は、幹細胞から分化した多様な種類の細胞から構成され、異なる役割を持つ細胞同士が協調的に働くことで、その機能を実現します。組織を構成する細胞の種類は、遺伝子の組み合わせで決定され、DNAやDNAに結合するタンパク質の化学修飾などの「エピゲノム」がその組み合わせ方を決めていると考えられています。大規模なエピゲノム解析は、国家プロジェクトレベルで世界中で進められ、ゲノムのわずか2%と言われる遺伝子に該当する領域の外にある非コード領域の機能的要素が次々と明らかになってきました。上記のプロジェクトでも採用され、業界のスタンダードとなっていたエピゲノム解析法ChIP-seq[用語3]は、解析に数百万〜数千万の多量の細胞数を必要とします。さらに、組織のエピゲノム解析では、組織が多様なタイプの細胞の混合物であるため、アンバランスなサンプリングを避けることができません。すなわち、幹細胞など数が限られた、しかし関心の高い細胞の情報は、他の多数の細胞の情報に埋もれてしまいます。たとえ、近年発展の目覚ましい単一細胞解析であっても、細胞の単離(組織から剥がして分取)が必要なことがほとんどで、細胞のロスや、ストレスが避けられないなど、「全細胞を対象としたエピゲノム解析」は非常にハードルの高い課題でした。
【研究の内容】
本研究グループは、同グループが2019年に発表した少数細胞エピゲノム解析法ChIL-seqを新たに組織のエピゲノム解析法として改良を行い、遺伝子発現の制御因子(RNA Polymerase II [用語4] やヒストン修飾 [用語1])の移動度を評価するデータ解析手法を組み合わせることで、組織中の埋もれた幹細胞の活性化情報(エピゲノム情報の変化)を検出する新たな解析技術を開発しました。提案した方法は、一辺3mm程度の小さな組織切片一枚(数千程度の細胞が含まれる)を使って、組織全体のエンハンサー、転写因子[用語5]、および転写が活性化した遺伝子の同定に十分な感度、特異性、再現性を持つことを示しました。組織に特化したChIL-seqは、蛍光付き抗体による組織中のタンパク質局在の可視化といったマイクロスケールの解析と、ChIP-seqより少ない細胞数(1/10,000程度)を用いて、ナノスケール(遺伝子解像度)で全ゲノム領域を対象とする網羅的解析が両立できる随一の解析手法と言えます。
従来の遺伝子発現やエピゲノムデータ解析は、ゲノム上に検出されるシグナルの量(高さ)に注目した定量的な解析が多く行われてきました。高感度かつ高精度なChIL-seqのデータを活用することで、遺伝子周囲のシグナルの「量」と、右肩上がり/下がりなどシグナルの分布形状を表す「形」の情報がより詳細に評価できます。今回の論文では、これら「量」と「形」の情報を取り出す統計モデルを作ることで、組織に占める細胞集団のサイズと、遺伝子活性化の強度情報が個別に得られることを示しました(図1)。すなわち、組織の組成(集団サイズ)のバイアスを受けない遺伝子活性情報が得られることになります。
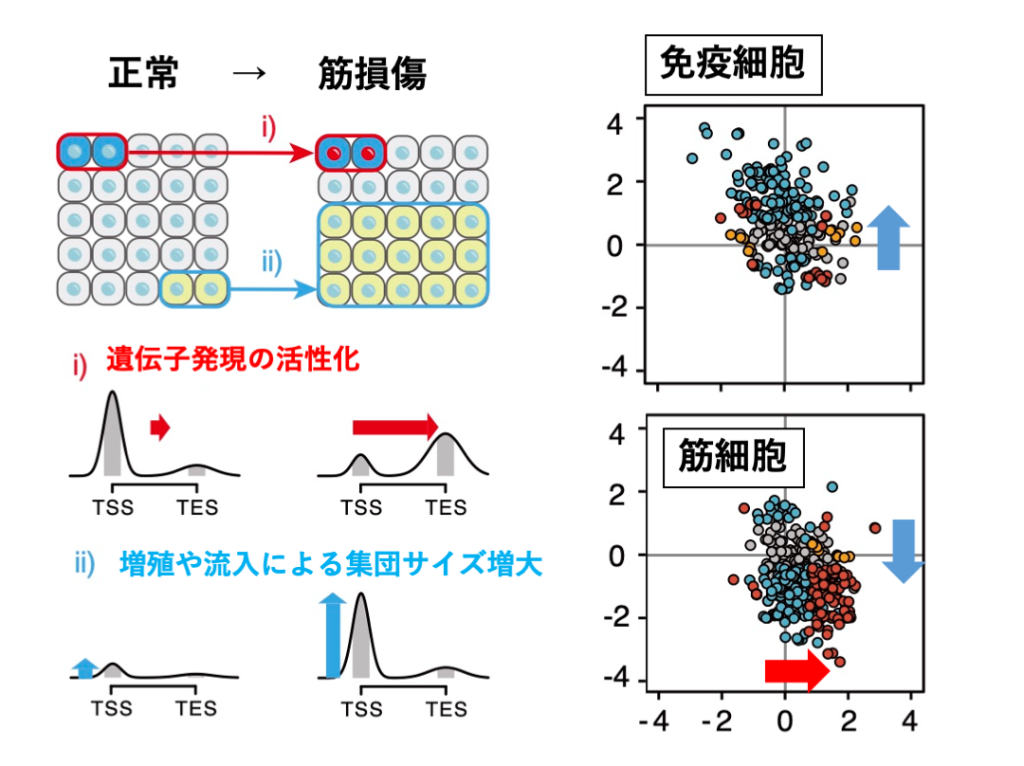
図 1:エピゲノムシグナルの量と形の分離による組織中の細胞集団プロファイリング:シグナルの量と形の変化を見ることで、特定細胞集団の増減や遺伝子の転写活性化を見出すことができる。TSS、TESは遺伝子の先頭と末端(転写開始点と終了点)を示す。
さらに、本解析法は、希少なため多量の細胞の入手が難しく、細胞の組成が多様なため解析の難しいがん組織(乳がん)のプロファイリングにも有用であることを示すことができました(図2)。
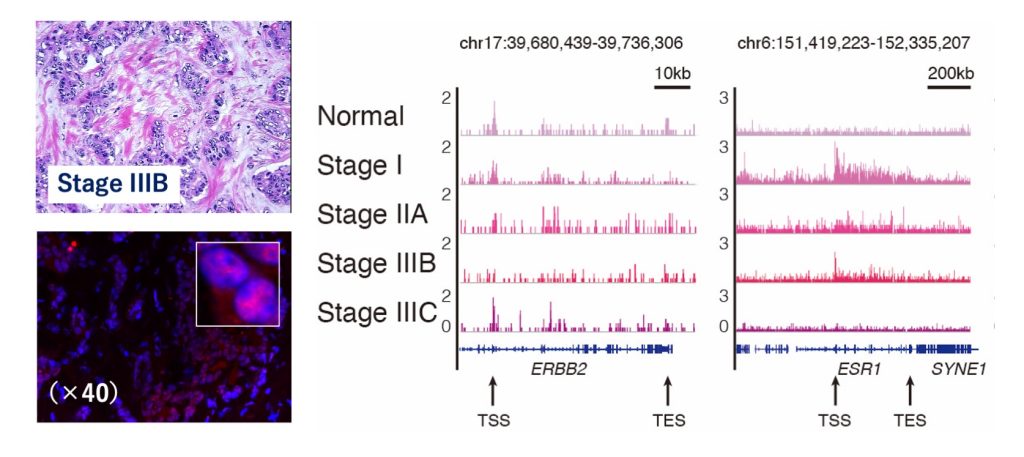 図 2:ChIL-seqによる乳がん組織のプロファイリング:ステージIIBの乳がん組織切片(左上、Origene社より購入)、RNA Polymerase II(赤)の切片上の局在(左下画像)、およびゲノム上のRNA Polymerase IIの局在(右側グラフ;代表的な乳がんマーカー遺伝子周囲)を示した。
図 2:ChIL-seqによる乳がん組織のプロファイリング:ステージIIBの乳がん組織切片(左上、Origene社より購入)、RNA Polymerase II(赤)の切片上の局在(左下画像)、およびゲノム上のRNA Polymerase IIの局在(右側グラフ;代表的な乳がんマーカー遺伝子周囲)を示した。
【今後の展開】
ChIL-seqは、これまで組織を対象としたエピゲノム解析の標準的な手法であったChIP-seqに代わるものとして大きな可能性を秘めています。組織切片上のタンパク質の局在から分かる空間情報と、ゲノムワイドな情報を組み合わせることで、分子〜細胞スケールの局所的相互作用の全体である組織の謎に迫ることができると考えています。現代は、情報技術と実験技術両者の相互発展の大きな潮流の只中にあります。この計測・数理科学の相互の発展を通して、新しい生命像や世界の捉え方が生まれることを期待しています。
【用語解説】
[用語1]
幹細胞:組織や器官を構成する分化した細胞の元となる細胞。多能性を持つ胚性幹細胞やiPS細胞などがよく知られているが、特定の細胞にのみ分化するような成体幹細胞も存在する。これらの幹細胞は存在量が少なく、その解析が難しい。
[用語2]
エピゲノム:後天的なゲノム制御情報。DNAの塩基配列に加えて、DNAそのものやDNAに強く結合するヒストンの化学修飾(ヒストン修飾)などにより、遺伝子の発現が制御される。エピゲノム情報を解読することにより、種々の細胞内で使われる遺伝子と使われない遺伝子がどのように区別されるか、調べることができる。
[用語3]
ChIP-seq(クロマチン免疫沈降シーケンス法):解析のターゲットとなるタンパク質が結合した位置の近くのDNA配列を取得することで、ゲノム中のタンパク質結合位置を網羅的に同定することができる方法。
[用語4]
RNA Polymerase II:遺伝子のDNA配列を読み取り、コピーであるmRNAを作る(転写する)ためのタンパク質。
[用語5]
転写因子:特定のDNA配列に結合し、遺伝子の発現を制御するタンパク質の総称。転写因子が結合するゲノム領域は、機能や制御される遺伝子からの距離に応じて、プロモーターやエンハンサーと呼び分けられている。